Legemidler basert på CRISPR
CRISPR (forkortelse for «clustered regularly interspaced short palindromic repeats») er en teknologi som gjør det mulig å gjøre spesifikke endringer i DNA i levende organismer. Verktøyet for genredigering gjør det mulig å klippe hvor som helst i genene til planter, dyr og mennesker. Slik kan man endre på DNA, «reparere» mutasjoner, slå ut gener og så videre [363].
De senere årene er det utviklet en del avanserte terapier basert på CRISPR/Cas9 (eller andre CRISPR-systemer) for å korrigere mutasjoner som forårsaker sykdom. Tabellen nedenfor gir en oversikt over legemidler med CRISPR som er vurdert av EMA. I de fleste tilfeller har legemidlene fått tildelt «Orphan designation» [364], en status som tildeles legemidler til bruk i behandling av sjeldne tilstander. For å få status som «orphan drug» må legemiddelet oppfylle nærmere gitte kriterier. «Orphan drugs»-klassifisering kan dra nytte av insentiver som beskytter mot konkurranse når det først er på markedet.
Årstall | Sykdom | Beskrivelse av legemiddelet | Bruk av CRISPR | Godkjent for bruk |
|---|---|---|---|---|
2019 | Beta-thalassemi er en arvelig sykdom der pasienter ikke klarer å produsere nok hemoglobin, proteinet som finnes i røde blodceller og frakter oksygen rundt i kroppen. Beta-thalassemi major er en alvorlig form av sykdommen der pasienter trenger hyppige blodtransfusjoner, mens beta-thalassemi intermedia er en mindre alvorlig form som kan forverres med alderen. Begge typer beta-thalassemi skyldes defekter i genet som er ansvarlig for produksjonen av beta-globin. | Casgevy består av umodne beinmargsceller (hematopoietiske celler) som tas fra pasienten. Cellene modifiseres for å få dem til å produsere gamma-globin, en av komponentene i føtalt hemoglobin. Når cellene gis tilbake til pasienten, forventes de modifiserte cellene å produsere gamma-globin, som igjen vil føre til produksjon av føtalt hemoglobin. Dette forventes å øke dannelsen av nye røde blodceller og redusere anemi. | Modifikasjonen av cellene gjøres ved hjelp av CRISPR-cas9, et enzym kombinert med et lite stykke genetisk materiale (RNA) som er i stand til å redigere et spesifikt gen. I dette legemidlet skaper CRISPR-cas9 defekter i et gen for et protein kalt BCL11A, som normalt stopper produksjonen av gamma-globin. Disse defektene hindrer produksjonen av BCL11A og tillater gamma-globin å bli produsert. | Ja [366], i 2024 |
2020 | Sigdcellesykdom/anemi er en alvorlig genetisk sykdom der de røde blodcellene endrer form fra å være skiveformede til å bli halvmåneformede (som en sigd). Formendringen skyldes en unormal form for hemoglobin. Hos pasienter med sigdcellesykdom fester de unormale røde blodcellene seg til veggene i blodårene og blokkerer dem, noe som begrenser strømmen av oksygenrikt blod til indre organer som hjerte, lunger og milt. Sykdommen forårsaker sterke smerter og skade på disse organene, samt gjentatte infeksjoner og anemi (lavt antall røde blodceller).
| Casgevy, se over | Se over | Ja, i 2024, se over |
Orphan drug-status, men ikke autorisert for bruk utenfor kliniske studier | ||||
Årstall | Sykdom | Beskrivelse av legemiddelet | Bruk av CRISPR | Status |
2020 | Epidermolysis bullosa [367] er en gruppe arvelige sykdommer der huden er svært skjør og danner blemmer etter selv mindre friksjon (gnidning) eller skade. I de fleste tilfeller vises symptomer på epidermolysis bullosa fra fødselen, men for noen former oppstår ikke symptomene før i voksen alder. Sykdommene skyldes mutasjoner (endringer) i genene som er ansvarlige for produksjonen av visse proteiner som gjør huden sterk og elastisk, som kollagen eller keratiner. | Legemidlet er et hudtransplantat som inneholder pasientens egne celler med et gen som er «redigert» slik at cellene kan produsere et hudprotein (kalt kollagen 7) som tidligere ikke ble produsert riktig. Hudtransplantatet forventes å oppføre seg som sunn hud og ikke lenger danne blemmer eller forårsake andre symptomer på tilstanden. | Modifikasjonen av cellene gjøres ved hjelp av CRISPR-Cas9, en metode for genredigering som bruker et enzym kombinert med et lite stykke genetisk materiale (RNA), som tillater presis modifikasjon av det unormale genet (COL7A1). | Da søknaden ble sendt, var det ikke startet noen kliniske studier på pasienter med epidermolysis bullosa. Legemiddelet var ikke godkjent for bruk i behandling. COMP ga en positiv uttalelse den 22. januar 2020, og anbefalte å gi status som «orphan drug». |
2022 | Pyruvatkinasemangel er en sjelden, arvelig metabolsk sykdom som påvirker enzymet pyruvatkinase. Tilstanden skyldes mutasjon i PKLR-genet. Klinisk lider pasienter med PK-mangel av en svært variabel grad av kronisk hemolyse, som spenner fra alvorlig neonatal gulsott og fatal anemi ved fødselen, alvorlig transfusjonsavhengig kronisk hemolyse, moderat hemolyse med forverring under infeksjon, til fullt kompensert hemolyse uten tilsynelatende anemi. Symptomer er vanligvis til stede ved fødselen, men på grunn av den varierende alvorlighetsgraden kan det ta tid før symptomene gjenkjennes [368]. | Legemidlet består av et virus som inneholder en fungerende kopi av PKLR-genet, som er ansvarlig for å lage pyruvatkinase-enzymet og mangler hos pasienter med pyruvatkinasemangel. Umodne benmargsceller med evne til å utvikle seg til røde blodceller tas fra pasienten, og viruset brukes til å levere genet inn i disse cellene. En permanent modifikasjon av cellene gjøres ved hjelp av den andre komponenten av legemidlet, som kalles CRISPR-Cas9. Når de modifiserte cellene transplanteres tilbake til pasienten, forventes de å produsere røde blodceller som kan produsere det manglende enzymet. | Autologe CD34+ celler redigert med CRISPR/Cas9 og transdusert med en adeno-assosiert virusvektor serotype 6 som inneholder den kodon-optimaliserte versjonen av PKLR-genet. | Status som «orphan drug» februar 2022 [369]. Kan tas i bruk i kliniske studier. |
2022 | Hyper IgM-syndromer [370] er en gruppe sjeldne lidelser der immunsystemet ikke fungerer som det skal. De klassifiseres som sjeldne primære immunsviktsykdommer, som er en gruppe lidelser preget av uregelmessigheter i celleutviklingen og/eller cellemodningsprosessen i immunsystemet. | Legemiddelet Telethon_018, består av CD4 T-celler (en type hvite blodceller) tatt fra pasienten, som er modifisert for å bruke CD40LG-genet. Når cellene injiseres tilbake i pasienten, forventes de å samhandle riktig med B-celler (en annen type hvite blodceller). Dette forventes å forbedre antistoffnivåene. | Modifikasjonen av cellene gjøres ved hjelp av CRISPR-Cas9. CD4 T-cellene genredigeres i CD40KLG-lokuset. | Status som «orpghan drug» mai 2022 [371]. Kan tas i bruk i kliniske studier. |
2024 | MRCP2 duplikasjonssyndrom [372] er en sjelden genetisk nevrodevelopmental sykdom som skyldes en ekstra kopi (duplikasjon) av MECP2-genet. Tilstanden kan gi en rekke symptomer med varierende alvorlighetsgrad, inkludert lav muskeltonus (hypotoni), utviklingsforsinkelser, epilepsi, og nevrodevelopmental svekkelse som påvirker kognitive, motoriske og talefunksjoner. | Behandling for MRCP2 duplikasjonssyndrom. | Adeno-assosiert virus serotype 9 som inneholder CRISPR/Cas13Y og guide-RNA mot det menneskelige MECP2-genet. | Status som "orphan drug" mai 2024. |
2024 | Wiskott-Aldrich syndrom [373] er en sjelden X-bundet recessiv sykdom som påvirker immunsystemets funksjon. Tlstanden kan gi symptomer som eksem, trombocytopeni (lavt antall blodplater), immunsvikt og blodig diaré Pasienter med Wiskott-Aldrich syndrom har økt risiko for blødninger, hyppige infeksjoner og autoimmune lidelser. Sykdommen kan også føre til livstruende komplikasjoner og redusert forventet levealder.
| Behandling for Wiscott-Aldrich syndrom. | Autologe CD34+ celler redigert med et CRISPR/Cas9-system og transdusert med en adeno-assosiert vektor som inneholder en kodon-optimalisert versjon av WAS-genet. | Status som «orphan drug» mai 2024 [374]. Kan tas i bruk i kliniske studier. |
Om kliniske studier med genterapi [375]
I 2022 ble det innført en ny forordning for kliniske legemiddelutprøvinger i EU/EØS (forordning (EU) nr. 536/2014). I den forbindelse ble det opprettet en ny database hvor utprøvinger skal registreres og søkes godkjent. Tallene i figurene under er hentet fra denne databasen. Det er dessverre ikke mulig å skille på hvilken type avanserte terapier (ATMP) dette er, eller hvilken indikasjon utprøvingen er for. DMP sin erfaring er imidlertid at de fleste ATMPene er for behandling innen kreftområdet.
Figur 7‑1 Godkjente kliniske utprøvinger i EU/EØS der utprøvingslegemidlet er avansert terapi (ATMP) i perioden 1. januar 2022 til og med 7. mars 2025 fordelt på kommersielle og ikke-kommersielle søkere.
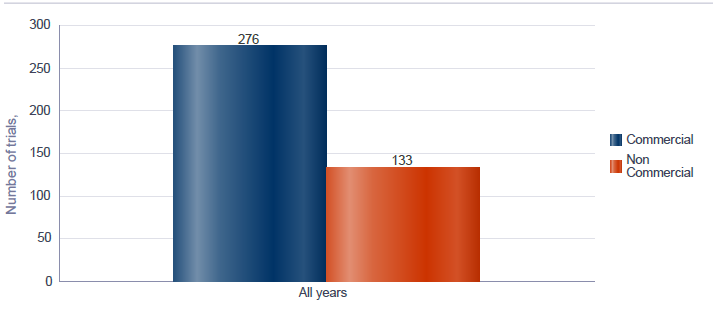
Forklaring til figuren: Figuren viser at det i perioden 1. januar 2022 til og med 7. mars 2025 var til sammen 409 kliniske studier med avanserte terapier – ATMP – som var godkjent i EU/EØS. Av disse var 276 kommersielle studier.
Figur 7‑2 Figuren under viser antallet godkjente kliniske studier i EU/EØS fordelt på faser der utprøvingslegemidlet er avansert terapi (ATMP) i perioden 1. januar 2022 til og med 7. mars 2025
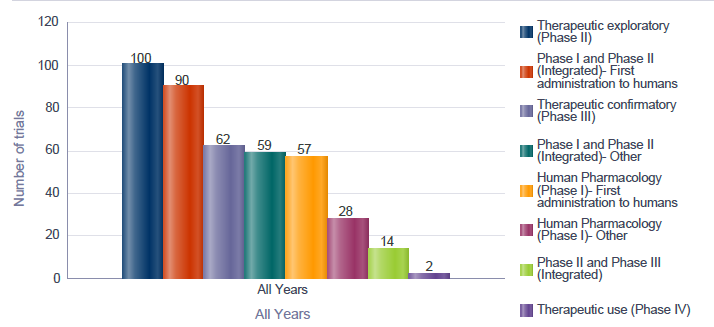
Forklaring til figuren: Figuren viser hvor mange studier som var hvilken fase av utprøvning. Det var for eksempel 100 studier i fase to, der legemiddelet prøves ut terapeutisk på mennesker, 90 studier i integrert fase en og to, der legemiddelet ble prøvd ut på mennesker for første gang og 62 studier i fase tre, der legemiddelet prøves ut terapeutisk til en større gruppe pasienter.
[363] Se for eksempel Et tiår med gensaksen CRISPR. – Det har vært en kjempeutvikling (forskning.no)
[365] Informasjon fra www.ema.europa.eu, med søk på CRISPR. Beskrivelse av sykdommer kan være hentet fra andre nettsider. Vi kan ikke garantere at vi har fått med alle aktuelle legemidler.
[367] Public summary of opinion on orphan designation Autologous skin equivalent graft composed of keratinocytes and fibroblasts genetically corrected by CRISPR/Cas9-mediated excision of mutation-carrying COL7A1 exon 80 for the treatment of epidermolysis bullosa (ema.europa.eu)
[375] Innspill fra spesialrådgiver Ingvild Aaløkken i Direktoratet for medisinske produkter.