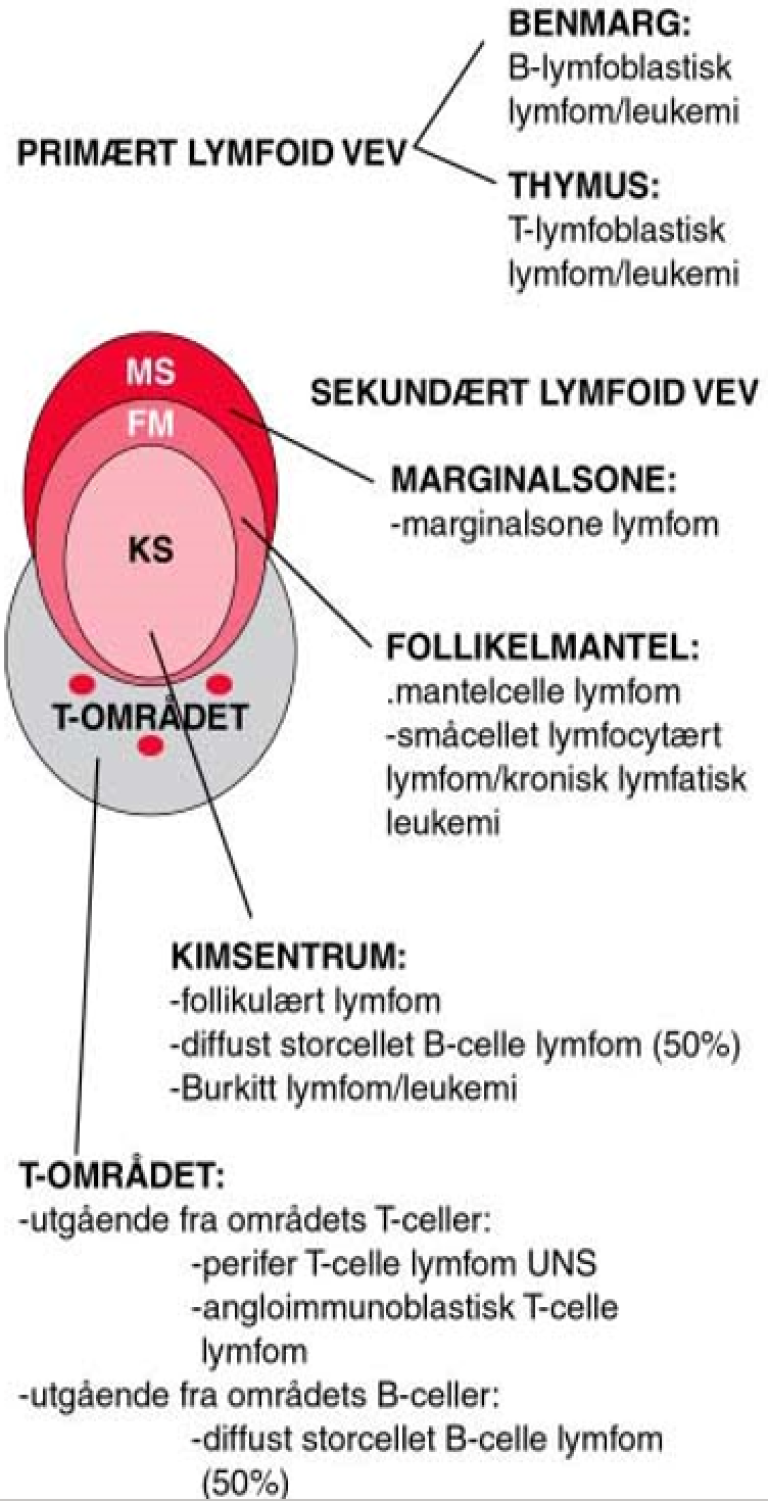
B-celle neoplasier kan betraktes som en klonal proliferasjon av B-celler «frosset» på et bestemt stadium av differensiering fra lymfoide stamceller til modne B-celler (hukommelses B-celler eller plasmaceller). De forskjellige lymfomsykdommer avspeiler således differensieringsstadier av normale B-celler og danner et viktig grunnlag for de nyeste klassifikasjonene (Alaggio et al., 2022; Campo et al., 2022; Swerdlow et al., 2016). B-lymfocyttene har sitt opphav i umodne B-celleforstadier (B-lymfoblaster), som igjen har sitt opphav i en lymfoid stamcelle og dernest i den multipotente, hematopoietiske stamcelle i benmarg. B-cellerekken differensierer i benmargen til modne B-celler med immunglobulin på overflaten. På dette stadiet har B-cellen ikke vært utsatt for antigen, og den forlater benmargen. B-cellene modnes videre i perifere lymfeknuter nårde blir antigenstimulert i T-celle området. Etter antigen-stimulering vil en del av B-cellene umiddelbart differensiere til plasmaceller, mens en del vil migrere til kimsenteret hvor immunglobulin reseptoren vil bli «skreddersydd» til antigenet via ulike mekanismer, blant annet hypermutasjoner av det antigenbindende sete. Noen B-celler differensieres i marginalsonen uten å gå gjennom kimsenteret. Disse sistnevnteB-cellene er antagelig viktige når det gjelder den såkalte T-celle uavhengige immunresponsen (for eksempel ved non-protein antigener). Feil i differensieringsprosessene av vanlige B-celler er sannsynligvis vesentlige under utviklingen av lymfomer. Mange lymfomer har for eksempel translokasjoner som fører til transformasjon og som er viktige elementer i utvikling av malignitet. Tilstedeværelse av genområdet for immunglobulinkjeder ved bruddpunktene for for onkogene kromosomale translokasjoner og hyppige mutasjoner i spesielle gener har ført til teorier om at feil i rearrangering av immunglobulingenene (V, D og J-gener), isotype skifte og somatiske mutasjoner i kimsenteret kan forårsake slike genskader. Dette plasserer kimsenterreaskjonen som et sentralt utgangspunkt for utviklingen av mange typer lymfomer. For noen lymfomer er det påvist en sammenheng med kronisk antigen stimulering og utviklingen av lymfom. Dette gjelder for eksempel for kronisk lymfatisk leukemi (KLL) medt en påfallende stereotyp bruk av spesielle immunglobulin generog flere former for marginalsonelymfomer som er knyttet til autoimmune tilstander eller kroniske infeksjoner .
Se figur 1 over med de viktigste lymfom-entitetene og deres opphavssted. B-lymfoblastisk leukemi/lymfoblastisk lymfom er sannsynligvis forskjellige kliniske uttrykksformer av samme sykdom. Opphavet er umodne prekursor B-lymfoblaster i benmargen. De maligne cellene ved en stor andel av mantelcellelymfomer er CD5 positive og korresponderer trolig mednaive B-celler som sirkulerer i blod og lymfe og finnes i mantelsonene i primære lymfoide follikler. Mantelcellelymfomer karakteriseres ved translokasjonen t(11;14) der regulerende deler av immunglobulingenet på kromosom 14 flyttes til kromosom 11 og gir overekspresjon av proliferasjonsproteinet cyclin D1. Dette igjen er sannsynligvis viktig for malign transformasjon av disse normale CD5-positive B-cellene. Mantelcellelymfomer kjennetegnes, som deres normale opphav, av at de ofte er småcellete, klinisk utbredte og gjerne leukemiske.
Også de maligne cellene ved KLL er også nesten alltid CD5 positive. Der immunglobulin tungkjede genet ikke er mutert, antas utgangspunktet å være normale CD5 positive B-celler uten kontakt med kimsenteret I om lag halvparten av tilfellene har immunglobulin tungkjede-genet gjennomgått en somatisk hypermutasjon som i en normal B-celle knyttes til interaksjon med antigen. Dessuten viser genekspresjons-undersøkelser at disse cellene har en genprofil som hukommelses B-celler. B-KLL er ikke karakterisert ved forekomst av et spesifikt gen-avvik som for eksempel mantelcelle lymfom og follikulært lymfom. Men noen genetiske avvik forekommer hyppig (del 13q, del 17p, del 11q og trisomi 12) og disse har sammenheng med mutasjonsstatus og prognostisk betydning. Man kjenner i liten grad årsaken til KLL, bortsett fra arv; risikoen for sykdommen er betydelig økt der førstegradsslektning har KLL eller annen lymfoproliferativ sykdom. Det foreligger dessuten store etniske forskjeller i insidens. Man tror KLL og SLL (småcellet lymfocytært lymfom) er forskjellige uttrykksformer av samme sykdom og at patogenese og etiologi derfor er lik.
Storcellete diffuse B-celle lymfomer og Burkitt lymfomer har sitt opphav fra blastiske celler som har vært stimulert av antigen i kimsentre og karakteriseres ved muterte immunglobulin V-gener. Diffuse storcellete B-celle lymfomer ofte også mutert BCL6 gen. Burkitt lymfom uttrykker Bcl-6 og har enten t(8;14), t(2;8) eller t(8;22) translokasjon som alle fører til overekspresjon av det proliferasjon-assosierte proteinet myc. Diffuse storcellete B-cellelymfomer kan også ha c-myc-translokasjon, men genekspresjonsarray kan likevel skille disse lymfomene fra Burkitt lymfom ved ulik ekspresjon av cellesyklusavhengige gener. Follikulære B-celle lymfomer har fenotype som centrocytter (modne centroblaster). Neoplasi oppstår sannsynligvis fordi centrocytter i kimsentre unngår apoptose som en følge av en kromosomal rearrangering t(14;18) som oppregulerer Bcl-2-ekspresjon. I motsetning til normale centrocytter, der ekspresjonen av det antiapoptotiske proteinet Bcl-2 nedreguleres, vil dette nå være vedvarende uttrykt. Marginal-sone B-celle lymfomer derimot, korresponderer til hukommelses B-celler med opphav i marginalsonen i follikler. Translokasjonen t(11;18), som man finner ved disse lymfomene, særlig ved lokalisasjon i ventrikkel, gir opphav til et fusjonsprotein og resulterer i overekspresjon av anti-apoptose proteinet API2. Det er usikkert hvorvidt lymfoplasmacyttisk lymfom også har sin opprinnelse fra marginalsone B-celler.
Infeksiøse agens har vært vist å kunne medvirke til utvikling av forskjellige former for B-celle lymfom (Ekström-Smedby, 2006; Suarez et al., 2006). Det mest klassiske eksempel er assosiasjonen mellom EBV og endemisk Burkitt lymfom i Afrika. Det er videre holdepunkter for at humant herpesvirus 8 spiller en viktig rolle ved primært effusjonslymfom og HIV-infeksjon ved multisentrisk Castlemans sykdom. Hepatitt C kan være assosiert med flere former for lymfom. SV40 virus spesifikke sekvenser ses i 40 % av NHL. Det spekuleres om dette er introdusert gjennom SV40-kontaminasjon i poliovaksine. Det sikreste eksempel på at en bakterie kan representere en utløsende årsak er MALT-lymfomer i ventrikkel, der eradikasjon av Helicobacter pylori kan gi tumor regresjon. Den viktigste kjente risikofaktor for å utvikle B-celle neoplasier er forandringer i immunsystemet, enten ved immunsuppresjon eller autoimmunitet. Immunsuppresjon som følge av HIV er en viktig risikofaktor for den betydelige økte insidensen av NHL hos denne pasientgruppen. Såkalt post-transplantasjons-lymfoproliferative tilstander (oftest B-celle lymfomer) forekommer hos pasienter som er organtransplanterte og bruker immundempende medikamenter i den hensikt å forhindre «graft versus host»-sykdom og avstøting av transplantater. Her er sannsynligvis redusert immunstatus sammen med EBV viktig for patogenesen. Det er en overhyppighet av NHL blant pasienter med reumatoid artritt. Sannsynligvis spiller både kronisk B-celleaktivering som ledd i autoimmuniteten og bruk av immunosuppressive medikamenter en rolle. Det er også rapportert NHL assosiert med Sjøgrens syndrom og Hashimotos thyreoiditt. For øvrig vet man at det er en viss overhyppighet av B-celle lymfomer blant personer som har vært utsatt løsemidler, fargestoffer og pestisider over tid. Det er generelt en noe økt risiko for søsken av pasienter med lymfoproliferative tilstander som lymfatiske leukemier og lymfomer til å utvikle slike sykdommer selv, og forskjellige studier, inklusive tvillingstudier viser at risikogener i kimbanen foreligger (Cerhan et al., 2015). Felles oppvekstmiljø spiller nok også en rolle for økt risiko i søsken av pasienter med lymfomer. Til tross for at det forekommer en opphopning av maligne lymfomer i enkelte familier, er insidensen av lymfom lav, den familiære risiko moderat og det finnes ingen screening metode for tidlig diagnostikk av lymfom. Dette gjør at aktiv oppføling av familier av lymfompasienter per i dag ikke er anbefalt.